人类参考基因组
人类参考基因组
一、人类参考基因组的来源
1、人类基因组计划
1)2001年草图,绘制人类基因组图谱
2、数据库的名称
1)UCSC:hg19,hg38
2)NCBI:GRCH19,GRCH38
二、如何下载参考基因组
在 linux 中下载参考序列数据库:
1. hg38:wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
2. hg19:wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
# 下载时间会比较久,建议网速不好时候,用其他方法。
三、参考基因组的后续处理
1、对下载的基因组进行初步整理:
tar zvfx chromFa.tar.gz
# 与 chrNo.fa 无关的文件删除掉。(chrNo 指 chr1、chr2......chrX、chrY、chrM)
cat *.fa >> genome.fa
rm chr*.fa
# 即可获得人类参考基因组序列的所有染色体的汇总文件:genome.fa
2、用 bwa 软件,对 genome.fa 建立索引文件:
[bwa path] index genome.fa
# 构建索引后,会生成文件:hg19.fa.amb、hg19.fa.ann、hg19.fa.bwt、hg19.fa.pac 和 hg19.fa.sa
四、参考基因组的信息统计
1、染色体的长度统计,用 python 画个条形图。
答:请参见以下程序1。
2、染色体的 GC 含量,用 python 画个条形图。
答:请参见以下程序2。
五、.fai 的深入研究
1、如何得到 .fai 文件 ?
答:用 samtools 软件的 faidx 参数,可对参考基因组 .fa 文件进行索引,生成 .fai 文件。
例如:[samtools path] faidx input.fa
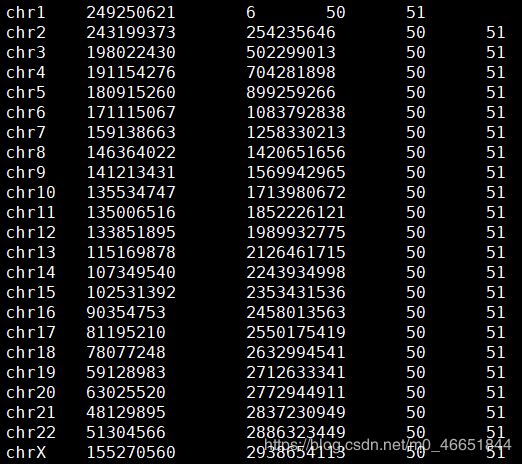
2、.fai 文件总共有 5 列信息:
1)序列所在染色体号
2)序列的长度
3)该序列的第一个碱基,在文件中的偏移位置,从 0 开始计数,first.base_offset
4)每行有多少个碱基, base_number_each_line
5)每行的字节长度, byte_each_line
例如:chr1 12345678 68 70 71
示例:
比如,2号染色体的第一个碱基的偏移量 = 1号染色体的长度/每行的碱基数 X 每行的字节数 + (>chr1\n 和 >chr2\n)的字节数。
3、如何利用 fai 文件快速得到某个 region 的参考序列 ?
答:用 samtools 软件的 faidx 参数,可快得到某个 region 的参考序列。
例如,如果要获得参考序列 hg19 的1号染色体上的 2377-2399 位置的序列:samtools faidx hg19.fa ’ chr1: 2377-2399 ’
4、给定一个 region,自己用 python 实现参考序列的快速查找 ?
答:利用 seek 函数,用 python 实现参考序列的快速查找。
请参见以下程序3。
程序1:
#!python3
# 读取genome.fa,进行染色体的长度统计,并用条形图呈现出来。
import sys
import matplotlib.pyplot as plt
import numpy as np
if len(sys.argv) != 2:
print('Usage: python3 %s ' % sys.argv[0])
sys.exit()
genome_fa = sys.argv[1]
fasta_open = open(genome_fa, "r")
l_hash = {}
# 染色体的长度统计,将染色体号和对应的长度,保存在字典中。
for line in fasta_open:
line = line.strip()
if line.startswith('>'):
chr_key = line.lstrip('>')
l_hash[chr_key] = 0
else:
l_hash[chr_key] += len(line)
fasta_open.close()
chrnum = list(l_hash.keys())
length = list(l_hash.values())
fig1 = plt.figure(num=1, figsize=(8,6))
plt.title('The length of each chromosome', fontweight='bold', fontsize=12)
plt.xlabel('Chromosome', fontsize=9)
plt.ylabel('Number of Base', fontsize=9)
plt.yscale('linear')
plt.xticks(rotation=45, fontsize=7)
plt.bar(chrnum, length)
plt.show()
fig1.savefig('Length_of_Chromosome.png')
程序2:
#!python3
# 读取 genome.fa,进行染色体的GC总含量的统计,并用条形图呈现出来。
import sys
import matplotlib.pyplot as plt
if len(sys.argv) != 2:
print('Usage: python3 %s ' % sys.argv[0])
sys.exit()
genome_fa = sys.argv[1]
fasta_open = open(genome_fa, "r")
b_hash = {}
# 染色体号为 key,GC 含量为 value,将统计数值保存在字典中。
for line in fasta_open:
line = line.strip()
if line.startswith('>'):
chr_key = line.lstrip('>')
b_hash[chr_key] = 0
else:
for base in line:
if base == 'C' or base == 'G':
b_hash[chr_key] += 1
fasta_open.close()
chrnum = list(b_hash.keys())
gc = list(b_hash.values())
# 绘制条形图。
fig1 = plt.figure(num=1, figsize=(8,6))
plt.title('The GC contents of each chromosome', fontweight='bold', fontsize=12)
plt.xlabel('Chromosome', fontsize=9)
plt.ylabel('GC Contents', fontsize=9)
plt.yscale('linear')
plt.xticks(rotation=45, fontsize=7)
plt.bar(chrnum, gc)
plt.show()
fig1.savefig('GC_Content_of_Chromosome.png')
程序3:
# 实现同 samtools faidx 一样功能的程序。
import sys
def Read_Base(filefa, offset, pos2, pos1):
seek = offset + pos1 - 1 # 该染色体第一个碱基偏移位置 + 序列的第一个碱基在染色体上的位置(-1是为了包括序列的第一个碱基,否则,会从序列的第二个碱基开始)
pos = pos2 - pos1 + 1 # 序列的长度。
with open(filefa, 'r') as fin: # 读 genome.fa,找序列的碱基段。
frontline = pos1 // 50 # 因为 genome.fa 是 50 个碱基为一行,看这条序列前面的碱基跨越了多少行。
remainline = pos1 % 50 # 看序列的第一个碱基在genome.fa中,是不是刚好在 \n 上(\n算是一个字节)
if remainline == 0: seek = seek + frontline - 2 # 如果在 \n 上,则指针要后移2个位置。
else: seek = seek - 1 # 如果不在 \n 上,指针只需要后移1个位置。
seekinitial = seek # 保存此时指针的位置,以防下面不满足规定时,需要回到这个位置上。
fin.seek(seek, 0) # 从头开始,看 seek 位置的碱基。
text = fin.read(pos + 1)
flag = True
while flag: # 循环程序目的:加上这条序列横跨的行,即\n的字节数。
if '\n' in text: # 如果该序列包括\n的话。
seek = seekinitial # 从原来的指针位置开始读。
count_n = text.count('\n') # 计算 \n 的数量。
seek = seek + count_n # 原来seek需要加上 \n 的字节数。
fin.seek(seek, 0)
text = fin.read(pos + 1)
if text.count('\n') <= count_n: # 如果seek加上原来\n的字节数后,又遇到\n,则再循环确保新的\n囊括进来。
flag = False
else: # 如果该序列不包括\n,则不用加\n的字节数。
flag = False
text = text.replace('\n','') # 去掉\n,只留下碱基。
lenbase = len(text)
baseline = lenbase // 60 # 模仿 samtools faidx 输出,每行为60个碱基。便于核对。
for l in range(baseline+1):
for t in text[0+60*l:60+60*l]: # 读取60个碱基,输出为同一行。
print(t, end='')
print('\n')
return
if __name__ == '__main__':
if len(sys.argv) != 6:
print('Usage: python3 run.py ')
sys.exit()
chrnum = sys.argv[3] # 读取哪一条染色体的序列。
num = chrnum.lstrip('chr')
# 如果是 chrX chrY chrM,转化为对应的数字。
if num == 'X':
num = 23
elif num == 'Y':
num = 24
elif num == 'M':
num = 25
num = int(num)
pos1 = int(sys.argv[4])
pos2 = int(sys.argv[5])
print('>{0}:{1}-{2}'.format(chrnum, pos1, pos2)) # 输出类似 >chrY:1000-1050 的信息。
with open(sys.argv[2], 'r') as f: # 读取 genome.fa.fai 文件。
for i in range(num): # 看看是哪一条染色体,则读取对应行的信息。
line = f.readline() # 获取想要读取的那一条染色体的信息。
linelst = line.strip().split('\t')
offset = int(linelst[2])
Read_Base(sys.argv[1], offset, pos2, pos1)
作者:m0_46651844