R语言使用cgdsr包获取TCGA数据示例详解
TCGA数据源
TCGA数据库探索工具
查看任意数据集的样本列表方式
选定数据形式及样本列表后获取感兴趣基因的信息,下载mRNA数据
选定样本列表获取临床信息
综合性获取
下载mRNA数据
获取病例列表的临床数据
从cBioPortal下载点突变信息
从cBioPortal下载拷贝数变异数据
把拷贝数及点突变信息结合画热图
TCGA数据源众所周知,TCGA数据库是目前最综合全面的癌症病人相关组学数据库,包括的测序数据有:
TCGA数据库探索工具DNA Sequencing
miRNA Sequencing
Protein Expression
mRNA Sequencing
Total RNA Sequencing
Array-based Expression
DNA Methylation
Copy Number
知名的肿瘤研究机构都有着自己的TCGA数据库探索工具,比如:
Broad Institute FireBrowse portal, The Broad Institute
cBioPortal for Cancer Genomics, Memorial Sloan-Kettering Cancer Center
TCGA Batch Effects, MD Anderson Cancer Center
Regulome Explorer, Institute for Systems Biology
Next-Generation Clustered Heat Maps, MD Anderson Cancer Center
其中cBioPortal更是被包装到R包里面
这里介绍如何使用R语言的cgdsr包来获取任意TCGA数据。
cgdsr包:R语言工具包,可以下载TCGA数据。
DT包:data.table包,简称DT包,是R语言中的数据可视化工具包。DT包可以将Javascript中的方法运用到R中,也能将矩阵或者数据表在网页中可视化为表格,以及其它的一些功能。
> setwd("C:/Users/YLAB/Documents/R/win-library/4.1/")
> install.packages("R.methodsS3_1.8.1.zip",repos=NULL)#安装
> install.packages("R.oo_1.24.0.zip",repos=NULL)#安装
> install.packages("data.table")
> BiocManager::install("cgdsr", force = TRUE)#安装
> library(cgdsr)
> library(DT)
#创建一个cgdsr对象
> mycgds <- CGDS("http://www.cbioportal.org/")
#检查下载是否成功,如果是FAILED就是没成功。
> test(mycgds)
getCancerStudies... OK
getCaseLists (1/2) ... OK
getCaseLists (2/2) ... OK
getGeneticProfiles (1/2) ... OK
getGeneticProfiles (2/2) ... OK
getClinicalData (1/1) ... OK
getProfileData (1/6) ... OK
getProfileData (2/6) ... OK
getProfileData (3/6) ... OK
getProfileData (4/6) ... OK
getProfileData (5/6) ... OK
getProfileData (6/6) ... OK
all_TCGA_studies <- getCancerStudies(mycgds)
> DT::datatable(all_TCGA_studies)
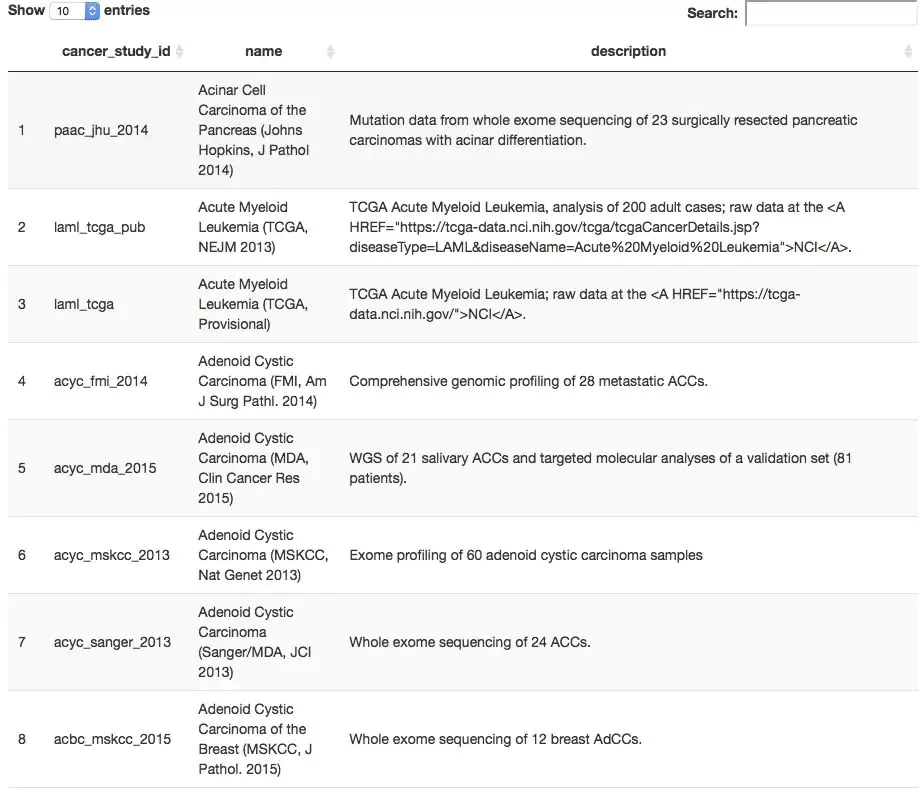
上表的cancer_study_id其实就是数据集的名字,我们任意选择一个数据集,比如stad_tcga_pub ,可以查看它里面有多少种样本列表方式。
stad2014 <- "stad_tcga_pub"
## 获取在stad2014数据集中有哪些表格(每个表格都是一个样本列表)
all_tables <- getCaseLists(mycgds, stad2014)
dim(all_tables) ## 共6种样本列表方式
[1] 6 5
DT::datatable(all_tables[,1:3])
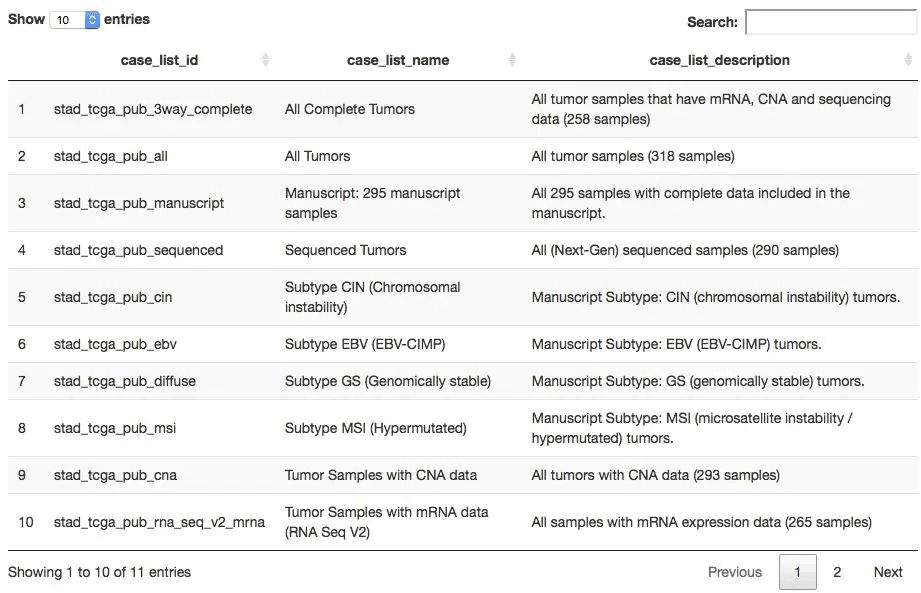
查看任意数据集的数据形式
## 而后获取可以下载哪几种数据,一般是mutation,CNV和表达量数据
all_dataset <- getGeneticProfiles(mycgds, stad2014)
DT::datatable(all_dataset,
extensions = 'FixedColumns',
options = list( #dom = 't',
scrollX = TRUE,
fixedColumns = TRUE
))
一般来说,TCGA的一个项目数据就几种,如下:
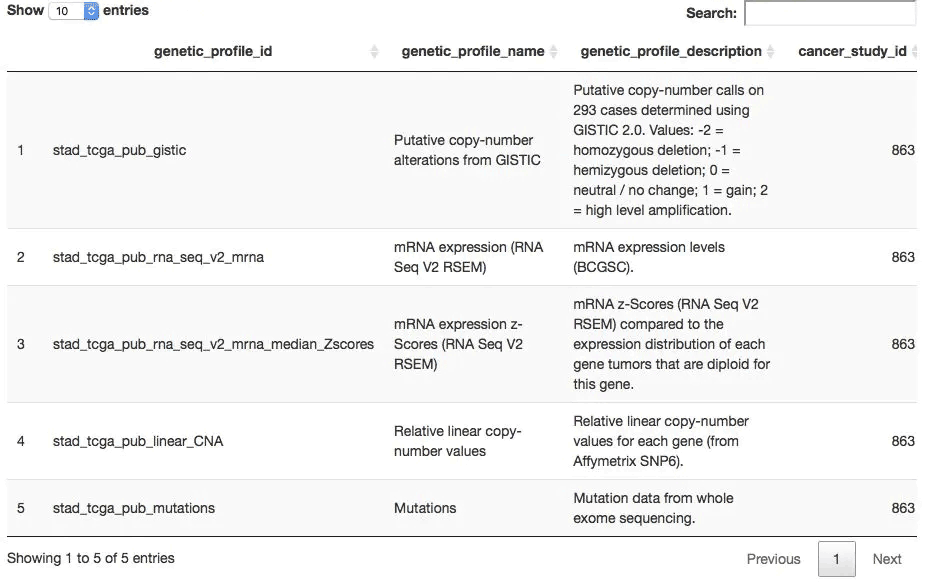
my_dataset <- 'stad_tcga_pub_rna_seq_v2_mrna'
my_table <- "stad_tcga_pub_rna_seq_v2_mrna"
BRCA1 <- getProfileData(mycgds, "BRCA1", my_dataset, my_table)
dim(BRCA1)
[1] 265 1
样本个数差异很大,不同癌症热度不一样。
## 如果我们需要绘制survival curve,那么需要获取clinical数据
clinicaldata <- getClinicalData(mycgds, my_table)
DT::datatable(clinicaldata,
extensions = 'FixedColumns',
options = list( #dom = 't',
scrollX = TRUE,
fixedColumns = TRUE
))
只需要根据癌症列表选择自己感兴趣的研究数据集即可,然后选择好感兴趣的数据形式及对应的样本量。就可以获取对应的信息:
library(cgdsr)
library(DT)
mycgds <- CGDS("http://www.cbioportal.org")
##mycancerstudy = getCancerStudies(mycgds)[25,1]
mycancerstudy = 'brca_tcga' getCaseLists(mycgds,mycancerstudy)[,1]
## [1] "brca_tcga_3way_complete" "brca_tcga_all"
## [3] "brca_tcga_protein_quantification" "brca_tcga_sequenced"
## [5] "brca_tcga_cna" "brca_tcga_methylation_hm27"
## [7] "brca_tcga_methylation_hm450" "brca_tcga_mrna"
## [9] "brca_tcga_rna_seq_v2_mrna" "brca_tcga_rppa"
## [11] "brca_tcga_cnaseq"
getGeneticProfiles(mycgds,mycancerstudy)[,1]
## [1] "brca_tcga_rppa"
## [2] "brca_tcga_rppa_Zscores"
## [3] "brca_tcga_protein_quantification"
## [4] "brca_tcga_protein_quantification_zscores"
## [5] "brca_tcga_gistic"
## [6] "brca_tcga_mrna"
## [7] "brca_tcga_mrna_median_Zscores"
## [8] "brca_tcga_rna_seq_v2_mrna"
## [9] "brca_tcga_rna_seq_v2_mrna_median_Zscores"
## [10] "brca_tcga_linear_CNA"
## [11] "brca_tcga_methylation_hm450"
## [12] "brca_tcga_mutations"
下载mRNA数据
mycaselist ='brca_tcga_rna_seq_v2_mrna'
mygeneticprofile = 'brca_tcga_rna_seq_v2_mrna'
# Get data slices for a specified list of genes, genetic profile and case list
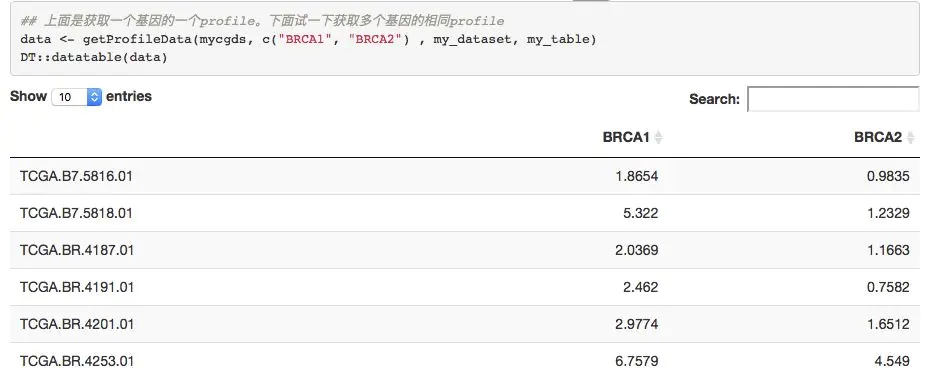
expr=getProfileData(mycgds,c('BRCA1','BRCA2'),mygeneticprofile,mycaselist)
DT::datatable(expr)
很简单就得到了指定基因在指定癌症的表达量
myclinicaldata = getClinicalData(mycgds,mycaselist)
DT::datatable(myclinicaldata,
extensions = 'FixedColumns',
options = list( #dom = 't',
scrollX = TRUE,
fixedColumns = TRUE
))
## Warning in instance$preRenderHook(instance): It seems your data is too
## big for client-side DataTables. You may consider server-side processing:
## http://rstudio.github.io/DT/server.html
从cBioPortal下载点突变信息
#突变基因名称集合
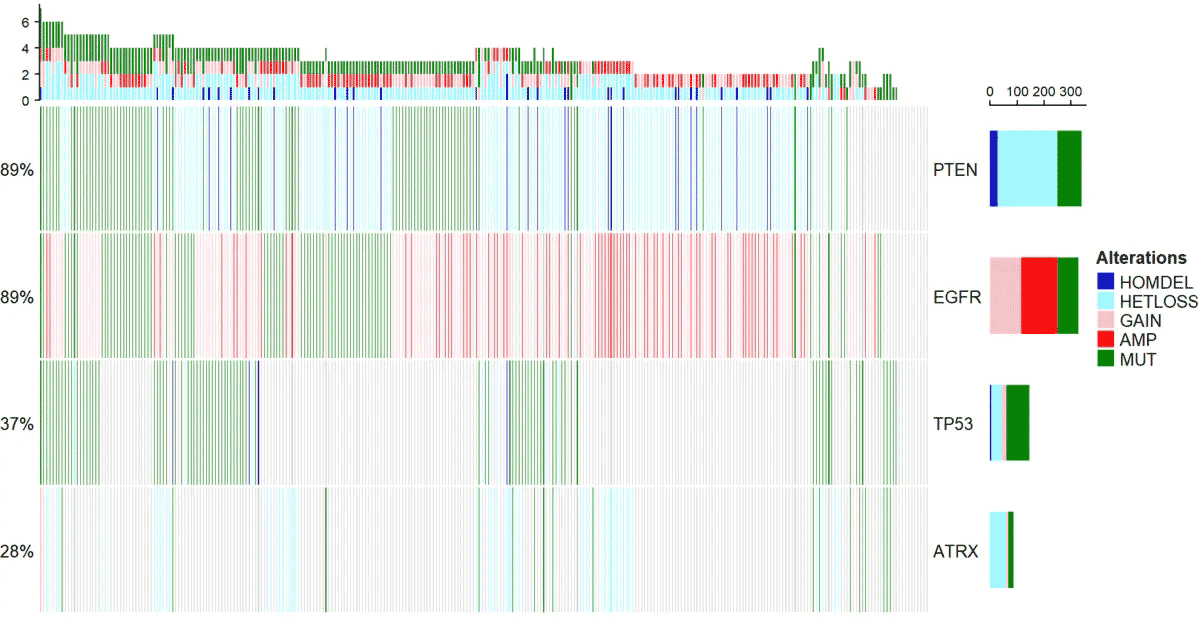
mutGene=c("EGFR", "PTEN", "TP53", "ATRX")
#检索基因和遗传图谱的基因组图谱数据
mut_df <- getProfileData(mycgds,
caseList ="gbm_tcga_sequenced",
geneticProfile = "gbm_tcga_mutations",
genes = mutGene
)
mut_df <- apply(mut_df,2,as.factor)
mut_df[mut_df == "NaN"] = ""
mut_df[is.na(mut_df)] = ""
mut_df[mut_df != ''] = "MUT"
DT::datatable(mut_df)
mutGene=c("TP53","UGT2B7","CYP3A4")
cna<-getProfileData(mycgds,mutGene,"gbm_tcga_gistic","gbm_tcga_sequenced")
cna<-apply(cna,2,function(x) as.character(factor(x,levels = c(-2:2),labels = c("HOMDEL","HETLOSS","DIPLOID","GAIN","AMP"))))
cna[is.na(cna)]=""
cna[cna=="DIPLOID"]=""
DT::datatable(cna)
下面的函数,主要是配色比较复杂,其实原理很简单,就是一个热图。
library(ComplexHeatmap)
library(grid)
conb <- data.frame(matrix(paste(as.matrix(cna),as.matrix(mut_df),sep = ";"), nrow=nrow(cna),ncol=ncol(cna), dimnames=list(row.names(mut_df),colnames(cna))))
mat <- as.matrix(t(conb))
DT::datatable((mat))
alt <- apply(mat,1,function(x)strsplit(x,";"))
alt <- unique(unlist(alt))
alt <- alt[which(alt !="")]
alt <-c("background",alt)
alter_fun = list( background = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="#CCCCCC",col=NA)) }, HOMDEL = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="blue3",col=NA)) }, HETLOSS = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="cadetblue1",col=NA)) }, GAIN = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="pink",col=NA)) }, AMP = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="red",col=NA)) }, MUT = function(x,y,w,h){ grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"), gp=gpar(fill="#008000",col=NA)) })
col <- c("MUT"="#008000","AMP"="red","HOMDEL"="blue3", "HETLOSS"="cadetblue1","GAIN"="pink")
alt = intersect(names(alter_fun),alt)
alt_fun_list <- alter_fun[alt]
col <- col[alt]
oncoPrint(mat=mat,alter_fun = alt_fun_list, get_type = function(x) strsplit(x,";")[[1]], col = col)
以上就是R语言使用cgdsr包获取TCGA数据示例详解的详细内容,更多关于R语言cgdsr获取TCGA数据的资料请关注软件开发网其它相关文章!